浆细胞样树突状细胞(pDC)是机体产生I型干扰素(IFN-I)能力最强的免疫细胞。它们由骨髓造血干细胞分化发育而来,成熟后经血流迁移至外周组织,通过内体上的TLR7/9识别病毒核酸,在数小时内迅速产生大量I型干扰素,启动抗病毒免疫应答。然而,这种“超能力”也是一把“双刃剑”。当pDC被自身核酸异常激活或者病毒过度激活时,过量产生的I型干扰素会引发系统性红斑狼疮或银屑病等自身免疫疾病。同时,在LCMV病毒感染中,pDC持续分泌的IFN-I甚至可导致病理性结肠炎。因此,pDC的激活和I型干扰素分泌必须受到精确调控。那么,机体如何确保pDC在“激活”与“过度激活”之间维持平衡呢?
2026年6月26日,清华大学药学院陈立功课题组联合清华基础医学院免疫所吴励课题组在Advanced Science在线发表题为“Slc44a2 deficiency unveils an IFN-I–dependent feedback control of pDC egress”的研究论文,揭示了pDC干扰素产生的反馈调控新机制:1)SLC44A2是IFN-I产生的重要负调控因子;2)骨髓pDC通过感应环境中的IFN-I信号,适时调整自身的迁出过程。这两种机制协同作用,共同维持pDC的稳态平衡,防止IFN-I反应的系统性过度激活。这一发现进一步完善了pDC负反馈调控的理论框架,表明pDC从发育分化、迁出到效应功能等环节均受到负反馈机制的系统性调控,为理解pDC在免疫应答中的稳态维持提供了新视角。同时,该研究为pDC相关疾病的干预提供了潜在新靶点。

01 | SLC44A2缺失导致pDC功能增强与骨髓滞留
研究团队通过系统筛选发现,溶质载体家族成员SLC44A2在静息pDC中高表达,激活后显著下调。为探究其功能,团队构建了Slc44a2基因敲除小鼠。结果发现:敲除Slc44a2后,骨髓中pDC数量正常,但脾脏、淋巴结、血液、肝脏等外周组织中pDC显著减少。进一步研究表明,这些pDC并非死于凋亡,只是被“困”在骨髓中无法向外周组织迁移,同时滞留的骨髓pDC呈现出显著的功能增强和活化标志物全面上调。为什么功能增强的pDC反而被困在了骨髓里?
02 | CCR2/CCR5下调介导pDC迁移受损
结合RNA-seq分析以及系统排查,研究人员发现敲除Slc44a2后,pDC表面两个关键的趋化因子受体CCR2和CCR5的表达量显著下调。CCR2和CCR5是引导pDC从骨髓向外周迁移的“导航仪”。当这两个受体减少,pDC便失去了离开骨髓的指令。遗传学实验进一步验证了这一机制:用抑制剂阻断CCR2/CCR5可导致外周pDC显著减少;敲除Ccr2或Ccr5基因同样导致外周pDC显著减少;双杂合小鼠的CCR2/CCR5表达水平与Slc44a2敲除小鼠相当,其外周pDC减少程度也高度相似。证实了SLC44A2缺失→CCR2/CCR5下调→pDC骨髓滞留。但SLC44A2缺失为何会导致CCR2/CCR5下调?
03 | IFN-I驱动的负反馈环:pDC的“自我封印”
研究团队注意到,敲除Slc44a2的pDC中I型干扰素信号通路异常激活。据此提出假设:过量的I型干扰素反过来抑制了CCR2和CCR5的表达。实验证实:CpG-A刺激pDC产生I型干扰素的同时,CCR2/CCR5持续下降;直接用IFN-α处理骨髓pDC可以显著下调CCR2/CCR5;用IFNAR1抗体阻断I型干扰素信号,CCR2/CCR5表达得以恢复;给野生型小鼠注射IFN-α,同样观察到骨髓pDC的CCR2/CCR5下调;在Slc44a2/Ifnar1双敲除小鼠中,CCR2/CCR5表达和外周pDC数量均显著恢复。
至此,一个完整的负反馈环路终于浮出水面:SLC44A2缺失→pDC功能增强→IFN-I过度产生→IFNAR信号激活→CCR2/CCR5下调→pDC骨髓滞留。当pDC产生过量I型干扰素时,I型干扰素反过来关闭pDC的“导航仪”,将pDC滞留在骨髓中,从而限制外周pDC的持续补充,防止系统性IFN-I反应过度激活——一个精妙的“自我封印”机制。
04 | 病毒感染中的负反馈:时间轴追踪
这是否是普遍存在的生理机制?研究团队利用LCMV-ARM病毒感染野生型小鼠,对感染后多个时间点进行时序分析。感染12小时后,血清IFN-α、骨髓中Sca-1+ pDC的比例及CD69的表达开始上升,骨髓pDC的CCR2和CCR5的表达及脾脏pDC比例开始下降;在24小时左右,血清IFN-α水平达到峰值;感染第7天后,血清IFN-α恢复至基线水平,脾脏pDC比例和CD69表达开始恢复;到第12天,CCR2/CCR5表达和Sca-1+ pDC的比例等指标基本恢复正常。时序上的精确对应揭示:LCMV感染诱导IFN-I产生,IFN-I通过IFNAR信号抑制CCR2/CCR5表达,阻断pDC迁出,限制外周pDC补充;当IFN-I回落,CCR2/CCR5恢复,pDC迁出重启。该反馈在感染早期抗病毒需求最迫切时启动,随病毒清除逐步解除,实现早期抗病毒应答与免疫稳态的动态平衡。
同样的现象在HSV-1感染和SLE模型中均得到验证,表明该负反馈机制是机体在多种炎症和自身免疫病理条件下广泛存在的pDC稳态调控系统。
05 | SLC44A2的转运底物
SLC44A2如何调控pDC的IFN-I产生?传统上SLC44A2被认为是胆碱和乙醇胺的转运蛋白,但团队发现敲除Slc44a2后,pDC内胆碱非但未下降,反而显著增加。同时,胆碱和乙醇胺并不影响pDC的I型干扰素产生,提示SLC44A2在pDC中可能存在其他关键底物。代谢组学显示,Slc44a2敲除后pDC内苏氨酸(T)、天冬酰胺(N)和谷氨酰胺(Q)显著积累。去除T/N/Q可显著抑制IFN-α分泌,补充则显著增强。分子对接进一步提示SLC44A2可结合并转运这三种氨基酸。这一研究首次将氨基酸转运代谢与pDC迁移调控联系起来,揭示了一个“代谢检查点——I型干扰素产生——细胞迁移”相互耦联的负反馈环路,为理解pDC稳态维持提供了全新视角。
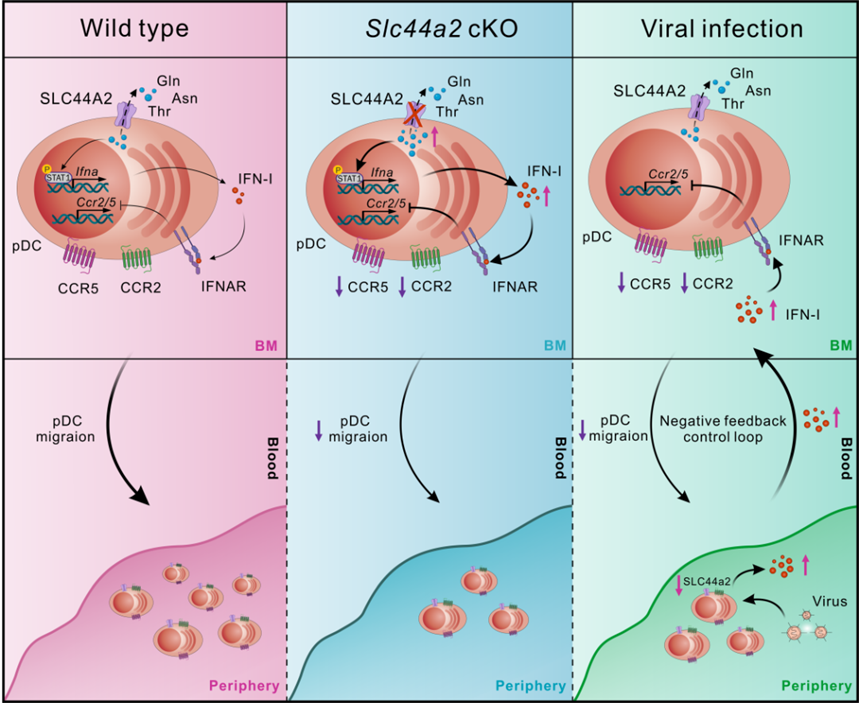
SLC44A2介导pDC稳态维持的机制图。该模型揭示了SLC44A2调控pDC稳态的两个核心机制:(1)SLC44A2可能通过外排氨基酸(T、N、Q)限制I型干扰素的产生,从而防止pDC自发激活。(2)环境中的I型干扰素信号通过下调CCR2和CCR5的表达,阻碍pDC从骨髓向外周组织的迁出。这两条途径协同作用,防止系统性I型干扰素过度反应,维持pDC的功能平衡。
清华大学药学院陈立功教授和基础医学院免疫所吴励教授为论文共同通讯作者。清华大学药学院已毕业博士陈瑞群、基础医学院及第一附属医院双聘人才吴韬博士为论文共同第一作者。 该研究获得国家自然科学基金、科技部国家重点研发计划、清华大学教育基金会、及清华-北大生命科学联合中心等多项基金的大力资助。
文章链接
https://doi.org/10.1002/advs.76325












 科研动态
科研动态