在临床感染治疗中,抗生素耐受性持久菌如同潜伏的“敌人”--它们不依赖耐药性基因突变,而是通过进入休眠状态逃避抗生素杀伤,成为耐药性进化的“储备库”,直接导致诸多感染治疗失败、病情反复。如何破解这一医学难题?
2026年1月2日,清华大学基础医学院刘佳峰课题组在国际期刊《科学进展》(Science Advances)发表研究成果,首次揭示细菌通过可逆的蛋白质凝聚体“开关”调控局部蛋白质合成,进而主动进入休眠状态以耐受抗生素的全新分子机制。这一发现为开发靶向持久菌的新型治疗策略、遏制抗生素耐药危机提供了关键理论支撑与潜在靶点。

刘佳峰课题组聚焦生命体休眠现象的进化原理及实现机制,尤其专注于细菌对抗生素耐受性的研究。此前研究虽提示蛋白质凝聚体可能与细菌休眠相关,但二者之间的具体调控逻辑一直是未解之谜。为攻克这一关键科学问题,课题组设计了循环抗生素压力进化实验,在对数生长期的大肠杆菌中施加间歇性抗生素压力,成功筛选出能在稳定期前提早停止生长进入休眠状态的耐受突变菌株。
深入分析发现,这些耐受菌株普遍携带丝氨酰-tRNA合成酶(SerRS)的基因突变。实验证实,无论是SerRS基因突变,还是使用丝氨酸类似物SHX(经典SerRS竞争性抑制剂),均会触发细菌因丝氨酸耗竭而进入生长停滞状态。更关键的是,课题组通过细胞成像技术观察到:突变SerRS蛋白会特异性“迁移”至由DEAD-box ATP酶(如DeaD)标记的蛋白质凝聚体中,而正常菌株的SerRS则均匀分布于细胞质,这一定位差异成为破解机制的核心线索。
进一步研究揭开了这一过程的动态调控规律:DeaD蛋白可通过液-液相分离形成凝聚体,且在体外实验中能优先富集突变蛋白SerRS。在细菌细胞内。另外,细菌体内DeaD标记的凝聚体本是活跃的蛋白质合成“热点区域”,但当突变的SerRS被募集进入后,细胞全局翻译水平正常而蛋白凝聚体内部的翻译活动显著下降--局部蛋白质合成的“暂停”,驱动细菌进入生长停滞和休眠状态;而一旦补充丝氨酸,SerRS便会从凝聚体中释放,局部翻译功能恢复,细菌随之重新启动生长。
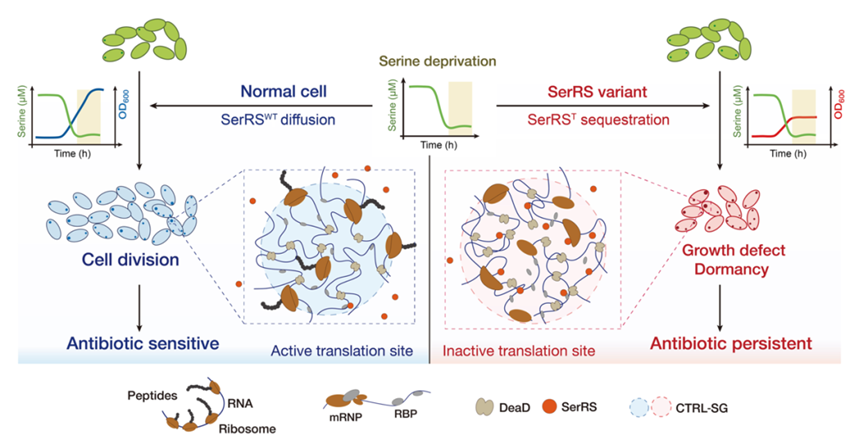
基于这一系列发现,课题组将这种响应丝氨酸水平、可精准调控细菌生长与休眠状态的蛋白质凝聚体结构,命名为“区室化翻译调控休眠开关颗粒”。这一“开关”的发现,不仅首次阐明了蛋白质凝聚体调控细菌持久性形成的具体分子路径,更打破了对细菌休眠机制的传统认知。
“持久菌的休眠并非被动的应激反应,而是由精准分子机制调控的主动适应策略”。该研究明确了蛋白质凝聚体作为“休眠开关”的核心功能,为靶向干预提供了明确方向--通过调控此类凝聚体的形成或解离,有望打破细菌的休眠保护,让抗生素重新发挥杀伤作用,从而降低耐药性进化风险。
刘佳峰课题组此前已在细菌耐受性与抗药性进化关系领域取得重要成果,此次新发现进一步拓展了对原核生物应激适应机制的理解。未来,课题组将继续围绕这一“休眠开关”展开深入研究,探索靶向该凝聚体的小分子化合物,推动基础研究成果向临床转化,为解决抗生素耐受这一全球性医学挑战贡献清华力量。
文章链接
https://doi.org/10.1126/sciadv.ady1930












 科研动态
科研动态